服务专业讲解
优势执行力高
价格合理优惠
特点量身定制
周期短
项目ce-mdr
地区全国
标准一站式全包服务
售后服务包售后
对于器械厂家是真正进入了MDR时代。对于已经取得MDD CE证书的企业,CE证书可以用到2024年5月26日。在2021年5月26日前尚未取得MDD 公告机构CE证书的企业,直接进入MDR 申请通道。对于普通一类的器械,2021年5月26日后需要申请MDR 欧盟注册,MDR 欧代,升级MDD技术文件为MDR完成MDR 符合性申明方可出口欧盟。
像轮椅,拐杖,护具,洗澡椅,座便器,助行器,矫正器,光学眼镜做MDR CE都是属于欧盟普通一类的产品。
按照MDD分类为I类的器械,绝大部分在MDR下仍然是I类器械。
一类器械的MDR 认证模式:
I类普通器械和MDD一样,仍然是走DOC模式;
I*类器械需要公告机构(MDR NB)参与认证、颁发证书。
MDR下的DOC和MDD的DOC不是一样的概念
从法规来说,I类普通器械也应有评估报告和上市后监督系统。
MDD可以将所有产品合并一本技术文件,MDR则需要进行分列,至少是预期用途完全一致的产品才可能共用一本技术文件;
从客户要求来说,由于MDR赋予进口商职责,因此欧洲买家会关注MDR合规性。
MDR法规下所谓的CE证书并不是,因为不是法规要求;
MDR下合规的是:
1)技术文件是否满足DRM的要求;
2)欧代是否按照MDR的要求进行了器械注册。
MDR法规对于普通I类没有提出认证要求;
MDR法规下,普通I类也不需要公告机构评审。
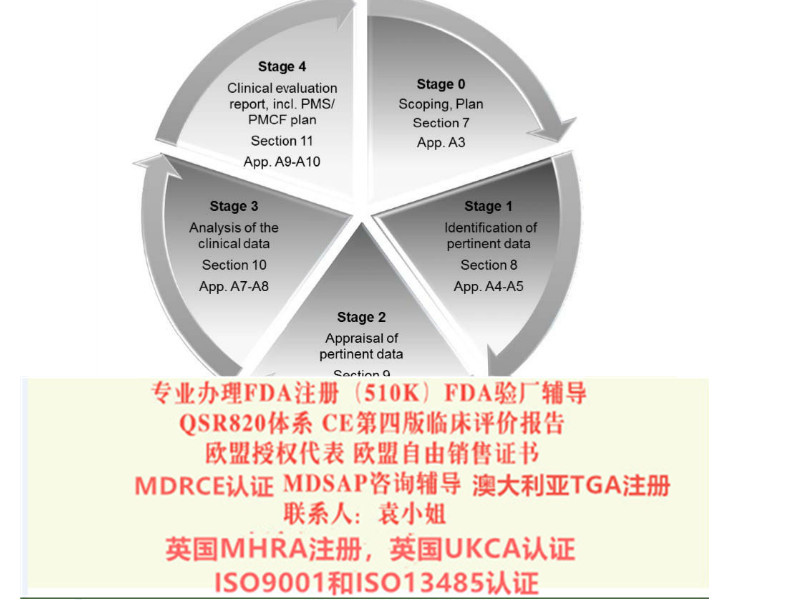
2017年5月5日欧盟就发布了新版器械法规MDR(EU 2017/745)。在2017年5月25日,MDR正式生效。老的器械指令即MDD( 93/42/EEC)与新的MDR(EU 2017/745)指令的交替过渡期为三年。
也就是说从2020年5月26日, MDR指令在欧盟就将开始强制执行,它将完全取代过去老的器械指令MDD (93/42/EEC)和老的有源植入器械指令AIMDD(90/385/EEC)。
需要特别指出的是:
☑ MDR强制执行后,新申请的CE认证必须按照MDR执行;
☑ 当前没有CE证书的产品,自5月27日起,必须按照MDR认证;
☑ 2020年5月26前签发的MDD证书,在有效期内仍然可以用,晚到2024年5月26日;
☑ 原有MDD证书需在证书失效前换发 MDR。
1. 本次爆发期间所获CE认证的医用口罩,可以说95%以上的都是按照老的MDD指令进行的,可能要面临新版换证问题;
2. 根据目前欧盟统计,拥有老指令版本MDD(93/42/EEC)授权的NB公告机构共有56家,而符合MDR授权的NB公告机构目前则仅有12家而已。也就是说,从2020年5月26日开始,针对医用口罩的CE认证审核机构可选性降低了80%;
3. 由于欧盟MDR此类授权审核机构(NB:Notified Body)可选性的减少,必然造成医用口罩CE认证费用相当大的概率将有大幅提升的可能;
4. 新版MDR指令审核要求比老版MDD指令更为复杂,认证周期必然大幅度拉长,本次期间有些机构声称的几天*的可能性将基本为零;
5. 获得CE认证的口罩等设备必须要求有的欧盟授权代表(简称"欧代"),这在以前MDD指令时对一些低风险产品其实有没有欧代并不严格,但在MDR指令后,即使是在一些电商平台上进行销售的产品也会要求提供必要的欧代信息;
6. MDR的体系审核流程和要求更为复杂与繁琐,举个例子如在MDR条款15中要求,器械厂商应在其组织架构内,至少配备一名负责合规的人员,即合规负责人(Person responsible for regulatory compliance)。该人员应具备器械领域的必要知识,并且有一系列的性要求(如法律、、、工程或其他相关学科,并且至少拥有一年与器械法规事务或质量管理体系相关的经验)。
7. 对于已经在欧盟渠道正式上市的产品来说,老的MDD指令CE证书可以保持到2024年5月26日。但如果企业产品在今年5月26日前,并未在欧盟市场销售的,原则上在今年5月26日后应该将老的MDD证书重新申请调整到MDR版本。

协调标准
标准和其他标准化出版物是自愿性准则,标准通常是由私人标准化组织在利益相关者的倡议下制定的,他们认为需要应用标准。尽管如此,但使用它们可以你的产品和服务达到一定的质量,*性和可靠性。
这里主要说下器械相关的(IVDD/MDD/AIMDD--MDR/IVDR)
器械指令Directive 93/42/EEC on Medical devices (MDD)
有源植入器械Directive 90/385/EEC on active implantable medical devices(AIMDD)
体外诊断试剂Directive 98/79/EC on in vitro diagnostic medical devices(IVDD)

给企业一些建议:
•要求的大幅增加对制造商(特别是中小企业)产生了巨大影响
•增加人才**性:制造商,机构,组,欧盟授权代表等需要熟悉法规,技术和人员
•尽早开始准备!
•检查产品分类和合规途径是否受到影响!
•内部自我检查,以确保技术文件:符合新的技术要求;特别是评估!满足语言要求
•了解MDR和内部差距分析的变化
•选择一个稳定,成熟的公告机构!
Medical Device Regulation 2017/745/EU法规是什么?
SUNGO集团凭借**网络和队伍为**客户提供法规,帮助企业*贸易壁垒,在器械行业尤为专长。
这主要包括:欧盟CE认证(MDD/MDR)、欧盟授权代表、器械欧盟注册、欧盟自由销售证书、FDA注册(FDA510K)、FDA验厂,陪审和翻译、ISO9001/ISO13485,中国局注册证、GMP体系和生产许可证等项目。
http://sungoyuan.b2b168.com







