周期4周
费用SUNGO
品牌SUNGO
欧代荷兰
注册CIBG注册
新的欧洲设备数据库eudamed将在提供数据和增加数据的数量和质量方面发挥核心作用(*33条)
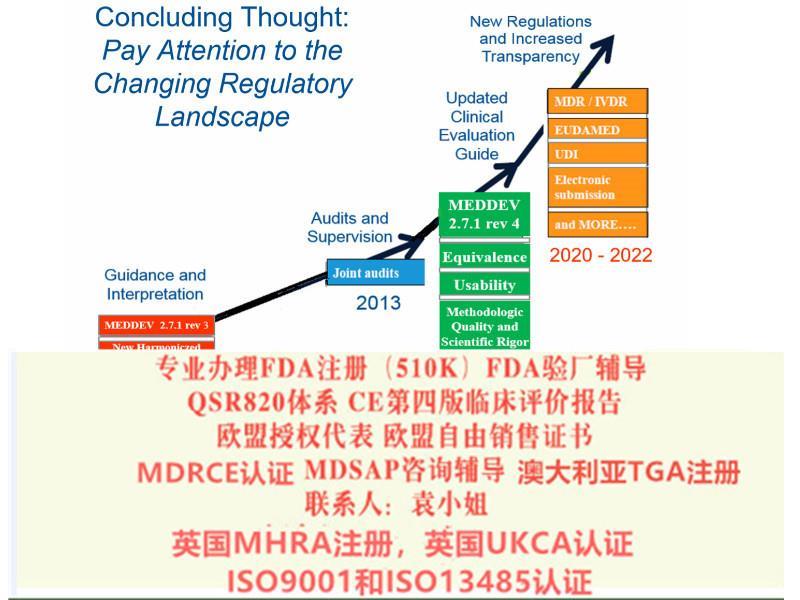
2021年MDR法规的实施日期逐渐临近,如何在过渡期截至前取得MDD的证书,如何或快速取得MDR证书。
说到这里 企业应该如何面对新法规的升级呢?按照MDR法规要求。关键的内容包括如下几个方面:
企业的质量管理体系 EN ISO13485:2016 ,产品的型式试验 TYPE TESTING , 产品的技术文件 TECHNICAL CONSTRUCTION FILES要满足这些要求,通常需要咨询机构和咨询师的协助。本公司拥有一支经验丰富的咨询师队伍可以为贵司提供这些服务,包括: 协助贵司建立/升级器械质量管理体系,将MDR法规的内容整合进去 协助贵司确定产品的欧盟协调标准,确认检测实验室的,样品准备以及检测不合格整改的研讨 按照MDR要求协助贵司准备技术文件,包括风险分析报告,评估资料,基本要求检查表等 协助贵司按照欧盟的要求修订说明书和标签,使其满足出口的要求。

申请欧盟CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
CE标志有何重要意义
CE标志的意义在于:用CE缩略词为符号表示加贴CE标志的产品符合有关欧洲指令规定的主要要求(Essential Requirements),并用以证实该产品已通过了相应的合格评定程序和/或制造商的合格声明,真正成为产品被允许进入欧共体市场销售的通行证。有关指令要求加贴CE标志的工业产品,没有CE标志的,不得上市销售,已加贴CE标志进入市场的产品,发现不符合要求的,要责令从市场收回,持续违反指令有关CE标志规定的,将被限制或禁止进入欧盟市场或退出市场。
CE是欧盟强制性的要求,所有销往欧盟市场的产品都必须标示“CE”,当然对作为救死扶伤的器械这一用具而言也不例外。在欧洲,除了主管当局如工商检查者将检查上市的器械是否带有CE标志,海关也将仅允许带有CE标志的产品通过边境。另外,器械的使用者(、)在购买新器械时也会检查是否带有CE标志。显然,CE标志可作为器械在欧盟内的“通行证”。同时,一个器械产品如果合法加贴了CE标志,也表明:
1、 该器械符合了欧盟器械法规的基本要求;
2、 该器械可以在欧盟市场内自由流通、销售及使用;
3、 该器械的整个形成过程已通过了一个相应的符合性评价程序。
欧盟器械CE指令
在器械领域,欧盟会制定发布了三个欧盟指令,以替代原来各成员的认可体系,使有关这类产品投放市场的规定协调一致。 这三个指令分别是:
1.基础器械指令(MDD,93/42/EEC),适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的器械,如无源性器械(敷料、一次性使用产品、接触镜、、导管等);以及有源性器械,如核磁共振仪、超声诊断和仪、输液泵等。该指令已于1995年1月1日生效,过渡截止日期为1998年6月13日,从1998年6月14日起强制执行。
2.体外诊断器械指令(IVDD,98/79/EC),适用于血细胞计数器,血糖仪、检测试纸、诊断、优生诊断等体外诊断用器械产品。
3.有源植入性器械指令(AIMDD, 90/335/EEC),适用于心脏起搏器,可植入的泵等有源植入性器械。AIMD于1993年1月1日生效。过渡截止期为1994年12月31日,从1995年1月1日强制实施。
上述指令规定,在指令正式实施后,只有带有CE标志的器械产品才能在欧盟市场上销售。

申请欧盟CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
Q:MDR何时生效?
A:2019年5月5日,欧盟正式发布了欧盟器械法规(MDR)。2019年5月25日,MDR正式生效。
器械指令MDD(93/42/EEC)和有源植入类器械指令AIMDD(90/385/EEC)被器械法规MDR(EU 2019/745)取代,法规过渡期为3年。
制造商应在过渡期内更新技术文件和流程以满足法规要求。具体可以参阅法规Article 120中若干过渡条款的要求。
法规中规定了对于一次性使用器械的再处理即复用的要求。
MDR
*17 条规定,一次性使用的器械的复用只能在相应国家法律允许的情况下进行,且应符合MDR 的规定。任何对一次性使用器械的再处理即复用的自然人或法人应视为复用器械的制造商,承担制造商义务,包括器械的可追溯性。但目前只有部分欧盟成员国接受器械复用并具备相应的法规规定。
MDR 在很多方面的规定都趋于更加严格的模式,更加强调持续和协作的方式。如从层面自上而下确定了欧盟、各成员国、公告机构、经济运营商各自的义务和责任,同时从法规层面设定了成员国之间、公告机构之间及制造商与部门之间沟通和协作的制度及途径,从产品角度来讲,从产品生产质量体系建立和实施、符合性评估过程中的通用基本要求、技术文件建立、上市后文件建立、证据等上市前要求,到符合性评估程序要求,以及上市后、警戒和市场等措施,覆盖产品生命周期的全过程,并规定了信息管理的具体要求,包括UDI 及市场的电子系统等。
基于本版法规的器械将很大程度上提高欧盟对器械产品的要求,不论是制造商还是公告机构都将面临更严格的管理,基于目前的产品分类规则,更多的产品将需要执行公告机构参与的符合性评估流程,更多的品种纳入了器械。
选择SUNGO,不是选择了一次性的合作伙伴,而是选择了一个长期的技术支持的战略伙伴。我们将秉承一贯“服务、客户”的原则,依托的技术团队,优化我们的服务,让更多的器械合法、进入市场,为器械行业健康发展贡献力量。
http://sungoyuan.b2b168.com








