-

-
上海沙格企业管理咨询有限公司
SUNGO Certification Company Limited - 13818104617


热门搜索:



起订量:1 价格:999
关于MDR涵盖产品范围和分类规则:器械的分类继续以前的类别,根据风险等级分为四类:I,II a,II b,III。但是,分类规则从18增加到22.具体分类规则的条件和变化如下:关于经济运营商的义务:该法规在章*2条的定义中提出了“经济运营者”的概念。

新法规将取代现行的三个器械指令:分别是器械指令93/42/EEC,有源器械指令90/385/EEC及体外诊断器械指令98/79/EEC。
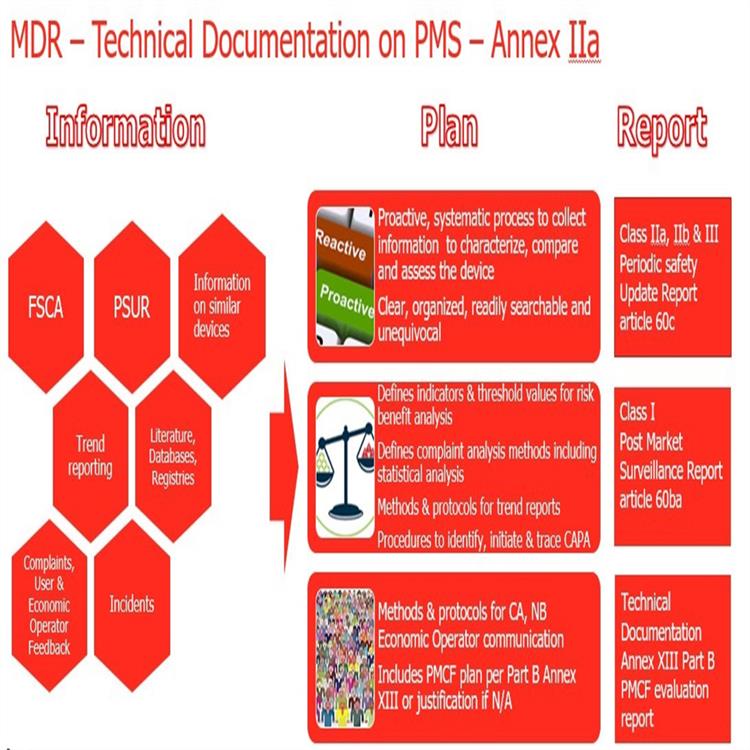
我司注意到新法规主要在以下几点上发生了变化:1.器械的定义;2.器械的分类;3.基本和性能要求;4.技术文件要求;5.评价;6.上市后;7.Eudamed数据库;8.对NB公告机构的要求(新法规生效后NB将按照新的要求重新进行授权);9.对高风险器械的新增了要求;MDR&IVDR修订要点15) 欧盟代表与进口商和制造商一起起到连带责任欧盟代表要求, 类似于法规负责人16) 平行贸 Parallel Trade, 特别是重新贴标签或重新包装17) 一次性器械的再加工和使用:- 符合成员国的法规要求- 再加工方要承担法定制造商的责任;- 由机构或外部加工方进行的一次性MD的再加工和再使用必须要符合通用技术规范或协调标准或成员国的法规要求.- 再加工器械的和性能应等同于初期的一次性MD18) 植入器械: 患者应获得基本的信息, 包括标识, 危害健康的警示或注意事项19) 带有CE标记的MD, 在欧盟内可以自由流通和销售. 但成员国可以限制某些器械的使用.20) 除了定制器械外, 所有器械都应应用UDI系统 (MDR执行后1-5年)21) EUDAMed, MD的 命名Code22) 植入和III类产品的制造商应公开产品的主要和性能, 及评估结果的概要.- 和性能的概要应特别包括该产品在与其他诊断或方法比较时的重要性,和二者的使用条件23) 在欧盟的层面上管理NB
