服务专业讲解
优势执行力高
价格合理优惠
特点量身定制
周期短
项目ce-mdr
地区全国
标准一站式全包服务
售后服务包售后
MDR法规下,I类器械(非测量、非、非重复使用)可以采用自我声明模式,但需要欧盟授权代表和欧洲注册。I类器械(测量、、重复使 用)除获得公告机构颁发的 CE和ISO13485证书外, 还需要欧盟授权代表和欧洲注册。
对于一个法定制造商而言,MDR*10条款所规定的所有一般性义务都是适用,另外MDR*52.7条款有对I类器械这些方面的要求进行规定。
I类(非无菌/非测量)器械基于MDD指令的“自我符合声明”,2021年5月26日之后是否依然合规?
基于MDD指令提供的自我符合声明,2021年5月26日起将不能合法投放欧盟市场。
I类无菌、带测量、可重复使用手术器械,是否需提供【定期*性更新报告PSUR】文档?
此类手术器械没有PSUR的要求。
但根据Art.85的要求,I类器械需提供上市后监督报告。

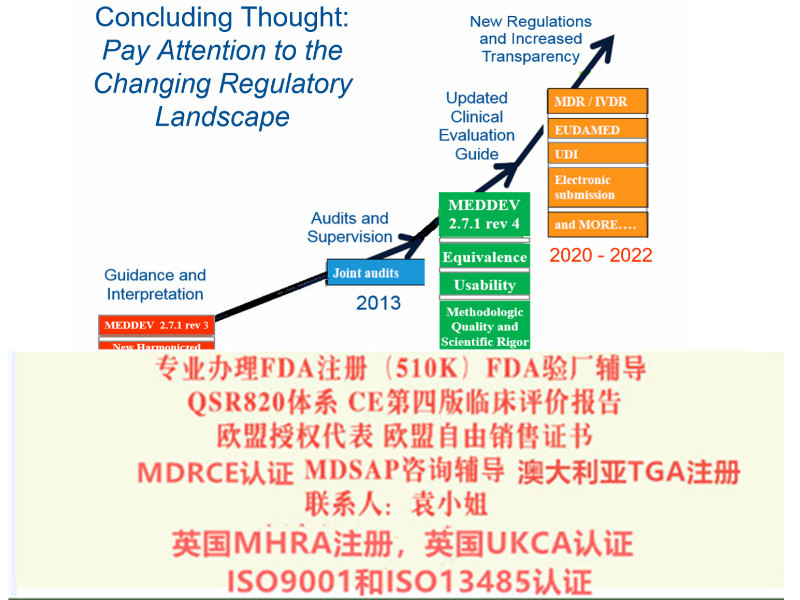
关于欧盟CE 认证的MDR法规升级:
老指令MDD 93/42/EEC including 2007/47/EC 升级到新法规MDR EU 2017/745
2017年5月,欧盟器械新法规MDR (REGULATION EU 2017/745) 颁布,新的法规将替代原有的器械指令 (MDD 93/42/EEC) 和有源植入性器械指令 (AIMDD 90/385/EEC) 。
从2020年5月开始公告机构不能按照MDD颁发CE证书,目前I及以上风险等级产品认证机构已不再受理MDD指令的认证申请。
对于目前获得CE证书的企业,应基于自身设备的证据的充分性合理安排申请MDR的时间,尽快启动MDR法规合规准备事宜
欧盟会规定了MDR 的转换期的要求
2017 年5 月25 日:MDR 和IVDR 生效
2020 年3 月25 日:启动欧盟器械数据库(Eudamed)
2020 年5 月25 日:MDR 实施开始
2022 年5 月25 日:IVDR 实施开始
2024 年5 月25 日:AIMD,MDD 和IVDD 证书将失效
作为器械制造商(生产商)产品在欧盟市场销售,需要做哪些工作呢?
制造商应做的事情:
在将产品投放市场之前准备技术文档
确保技术文档提供给市场当局(应要求看到它)只要产品投放市场
自产品投放市场之日起,将技术文档保存10年(除非另有明确规定)

对于器械厂家是真正进入了MDR时代。对于已经取得MDD CE证书的企业,CE证书可以用到2024年5月26日。在2021年5月26日前尚未取得MDD 公告机构CE证书的企业,直接进入MDR 申请通道。对于普通一类的器械,2021年5月26日后需要申请MDR 欧盟注册,MDR 欧代,升级MDD技术文件为MDR完成MDR 符合性申明方可出口欧盟。
像轮椅,拐杖,护具,洗澡椅,座便器,助行器,矫正器,光学眼镜做MDR CE都是属于欧盟普通一类的产品。
按照MDD分类为I类的器械,绝大部分在MDR下仍然是I类器械。
一类器械的MDR 认证模式:
I类普通器械和MDD一样,仍然是走DOC模式;
I*类器械需要公告机构(MDR NB)参与认证、颁发证书。
MDR下的DOC和MDD的DOC不是一样的概念
从法规来说,I类普通器械也应有评估报告和上市后监督系统。
MDD可以将所有产品合并一本技术文件,MDR则需要进行分列,至少是预期用途完全一致的产品才可能共用一本技术文件;
从客户要求来说,由于MDR赋予进口商职责,因此欧洲买家会关注MDR合规性。
MDR法规下所谓的CE证书并不是,因为不是法规要求;
MDR下合规的是:
1)技术文件是否满足DRM的要求;
2)欧代是否按照MDR的要求进行了器械注册。
MDR法规对于普通I类没有提出认证要求;
MDR法规下,普通I类也不需要公告机构评审。
http://sungoyuan.b2b168.com








