品牌SUNGO
欧代荷兰
周期4周
有效期5年
流程SUNGO
但是新MDR的管控趋于严格,对于制造商,欧盟授权代表以及国外进口商三方该承担的责任比较明确,欧盟授权代表和进口商与制造商一样为缺陷器械承担连带的法律责任。所以进口商在采购工厂产品的时候,较MDD老法规,他们更关注,工厂是否真正满足CE法规要求,尽量的将自己要承担的风险降低到。
SUNGO提醒我们的客户在申请产品CE认证时,在过渡阶段请谨慎考虑是选用法规还是采用老的指令方案,同时也需要对NB机构的发证进行了解和确认以保证产品在欧盟市场销售的可延续性。

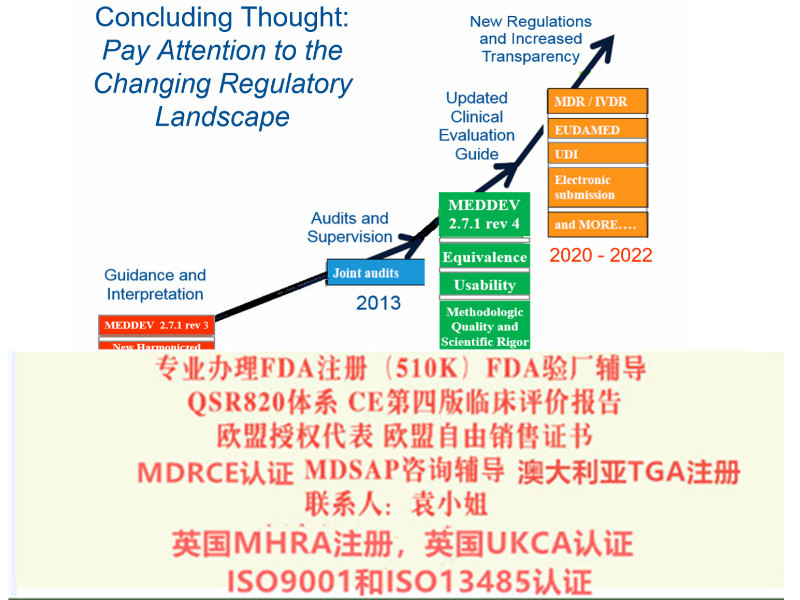
器械法规(MDR)转换期为3年,2020年5月4日起强制实行。体外诊断器械法规(IVDR)转换期为5年,2022年5月4日起强制实行
MDR框架
Chapter I – Scope and Definitions
Chapter II – CE Marking, Economic Operators, Reprocessing
Chapter III – Identification and Traceability of Devices
Chapter IV – Notified Bodies
Chapter V – Classification and Conformity Assessment
Chapter VI – Clinical Evaluation and Investigation
Chapter VII – Vigilance and Market Surveillance
Chapter VIII – Cooperation between Member States
Chapter IX – Confidentiality, Data Protection, Funding, Penalties
Chapter X – Final Provisions
100 whereas= Why?
X Chapters of 123 Articles = What?
XVI I Annexes = How?
I General safety and performance requirements
II Technical documentation
III Technical documentation on post-market surveillance
IV EU Declaration of conformity
V CE marking of conformity
VI European UDI System
VII Requirements to be met by notified bodies
VIII Classification rules
IX Conformity assessment based on a QMS and assessment of the technical documentation
X Conformity assessment based on type examination
XI Conformity assessment based on product conformity verification
XII Certificates issued by a notified body
XIII Procedure for custom-made devices
XIV Clinical evaluation and post-market clinical follow-up
XV Clinical investigations
XVI List of groups of products without an intended medical purpose
XVII Correlation table
内容 涉及
范围&定义 Chapter I
产品上市 Chapter II
UDI & 数据库 Chapter III, Annex VI,XII
NB Chapter IV , Annex VII,
符合性评估 Chapter V, Annex IX, X, XI
评价和调查 Chapter VI, Annex XIV ,XV
PMS, 警戒系统,上市后 Chapter VII , Annex XIV
Standards, timeline, MDCG Chapter VIII
Classification Annex VIII
一般性能要求(ER checklist) Annex I
TD要求 Annex II, III
DoC &CE marking Annex IV, V
Custom-made Annex XIII
非器械产品清单 & 指令、法规对比表 Annex XVI, XVII

类似于质量授权人注意到新法规主要在以下几点上发生了变化:1.器械的定义;2.器械的分类;3.基本和性能要求;4.技术文件要求;5.评价;6.上市后;7.Eudamed数据库;8.对NB公告机构的要求(新法规生效后NB将按照新的要求重新进行授权);9.对高风险器械的新增了要求;总的说来新的MDR和IVDR加强了体系管理,对高风险设备增加了相关规定比如对于非用途但具有与器械相似特性的设备也将受到新法规的管辖(如用于美容的彩色眼镜),提升了产品对患者的透明度和可追溯性并设立电子数据库(Eudamed)、设备将有一个的识别号这加强其在整个供应链的可追溯性。
我公司申请:ISO13485认证,CE评估报告编写 等产品出口的相关认证
http://sungoyuan.b2b168.com








