服务欧盟授权代表
代申请欧盟注册
申请CE认证
编写CE技术文件
建立ISO13485
在过渡期结束后,制造商是否仍然可以投放市场/投入使用符合指令的设备? 是的,在某些条件下,可以选择继续投放市场/投入使用符合指令的设备,直到其现有证到期为止。 这可以避免在MDR下立即需要新证。 要使用此选项,所有现有证必须有效(例如,QMS),设备的目的和性质不得更改,并且您必须遵循新的MDR规则进行注册,监视和警惕。
申请出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟自由销售证书、英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
2019年2月器械法规(MDR)和体外诊断器械法规(IVDR)终提案发布,2019年3月7日欧盟28个成员国一致表决同意欧盟采用新版的器械法规(MDR)和体外诊断器械法规(IVDR)。2019年5月5日,欧盟正式发布了OfficialJournal其正式对外宣布了新版MDR(REGULATIONEU2019/745)法规和新的IVDR(REGULATIONEU2019/746)法规。新法规将取代现行的三个器械指令:分别是器械指令93/42/EEC,有源器械指令90/385/EEC及体外诊断器械指令98/79/EEC。

至2020年5月26日,MDR法规将强制实施。至2022年5月26日IVDR法规将强制实施。至2014年,MDD/ AIMD证书全部失效。MDR新法规变化1)扩大了应用范围2)提出了新的概念和器械的定义3)细化了器械的分类4)完善了器械的通用和性能要求5)加强对技术文件的要求6)加强器械上市后的7)完善评价相关要求事故对于患者意味着伤害,对于企业的生存也具有巨大的破坏力。因此,评估的设计和数据的收集具有至关重要的意义。然而在实践中,许多制造商不清楚什么是欧盟法规所要求的评估,什么样的数据能满足欧盟的法规要求。

MDR框架
Chapter I – Scope and Definitions
Chapter II – CE Marking, Economic Operators, Reprocessing
Chapter III – Identification and Traceability of Devices
Chapter IV – Notified Bodies
Chapter V – Classification and Conformity Assessment
Chapter VI – Clinical Evaluation and Investigation
Chapter VII – Vigilance and Market Surveillance
Chapter VIII – Cooperation between Member States
Chapter IX – Confidentiality, Data Protection, Funding, Penalties
Chapter X – Final Provisions
100 whereas= Why?
X Chapters of 123 Articles = What?
XVI I Annexes = How?
I General safety and performance requirements
II Technical documentation
III Technical documentation on post-market surveillance
IV EU Declaration of conformity
V CE marking of conformity
VI European UDI System
VII Requirements to be met by notified bodies
VIII Classification rules
IX Conformity assessment based on a QMS and assessment of the technical documentation
X Conformity assessment based on type examination
XI Conformity assessment based on product conformity verification
XII Certificates issued by a notified body
XIII Procedure for custom-made devices
XIV Clinical evaluation and post-market clinical follow-up
XV Clinical investigations
XVI List of groups of products without an intended medical purpose
XVII Correlation table
内容 涉及
范围&定义 Chapter I
产品上市 Chapter II
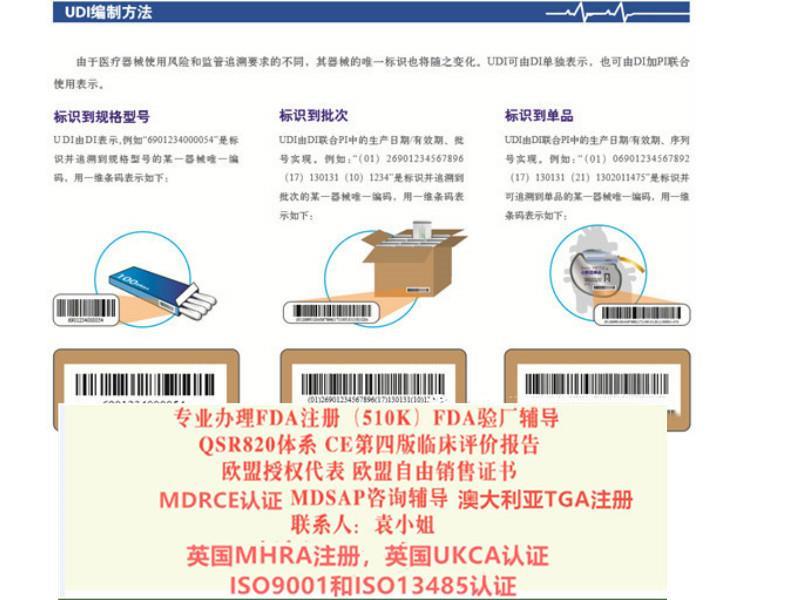
UDI & 数据库 Chapter III, Annex VI,XII
NB Chapter IV , Annex VII,
符合性评估 Chapter V, Annex IX, X, XI
评价和调查 Chapter VI, Annex XIV ,XV
PMS, 警戒系统,上市后 Chapter VII , Annex XIV
Standards, timeline, MDCG Chapter VIII
Classification Annex VIII
一般性能要求(ER checklist) Annex I
TD要求 Annex II, III
DoC &CE marking Annex IV, V
Custom-made Annex XIII
非器械产品清单 & 指令、法规对比表 Annex XVI, XVII
并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。对于特定Ⅲ类器械和Ⅱ b 类器械产品,在已委派必要的器械协调小组(MDCG)、小组前提下,同样可通过指令豁免在2020 年5 月26 日之前投放市场。法规关于公告机构的要求(正文*35~50 条) 自2017 年11 月26日起适用,即公告机构在新法规发布后的六个月内即应开始进行相应的申请,符合要求后方可依据新法规开展符合性评估。同时法规对成员国主管机构的和MDCG 的成立也设定了期限,要求于2017 年11 月26 日前完成。对于成员国主管机构之间的协调,设定期限为2018 年5 月26 日。
手术衣510K,隔离衣510K,手套510K,电动/手动轮椅510K,FDA注册,FDA美国代理人
http://sungoyuan.b2b168.com








