服务欧盟授权代表
代申请欧盟注册
申请CE认证
编写CE技术文件
建立ISO13485
新的标识识别系统(UDI系统)(*27条)将有力地增强市场后相关活动的可追溯性和有效性。
MDR还将提高透明度,公开有关设备和研究的信息。新的欧洲设备数据库eudamed将在提供数据和增加数据的数量和质量方面发挥核心作用(*33条)
2021年MDR法规的实施日期逐渐临近,如何在过渡期截至前取得MDD的证书,如何或快速取得MDR证书。上海沙格有着的咨询师团队,丰富的案例经验。
新MDR不仅包含了MDD及AIMDD涵盖的所有产品;还覆盖用于器械的清洁、或的器械,以及Annex XVI列举的无预期目的的产品,如美瞳、面部填充或、、皮肤改善和美容等产品。
包含某些药械结合产品,详细请见Article1(8,9)。
包含某些由非活性或处理为非活性的人类来源组织或细胞物制造的特定产品。
包含声称仅具有美容目的或另一种非目的,但在功能和风险特征方面类似于器械的特定产品组
声明纳米材料器械属于MDR范围,且要接受为严格的评估程序。
包含发射离子的器械和用途的软件。
MDR的主要变化
1.扩大了应用范围
2.提出了新的概念和器械的定义
3.细化了器械的分类
4.完善了器械的通用和性能要求
5.加强对技术文件的要求
6.加强器械上市后的
7.完善评价相关要求
8.提出Eudamed数据库的建立和使用
9.提出器械的可追溯性(UDI)
10.对NB提出严格的要求
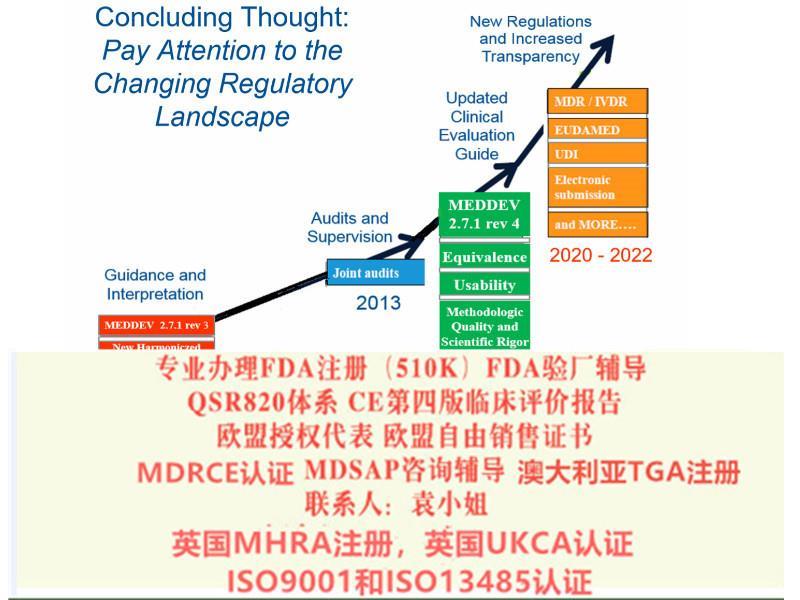
我司注意到新法规主要在以下几点上发生了变化:1.器械的定义;2.器械的分类;3.基本和性能要求;4.技术文件要求;5.评价;6.上市后;7.Eudamed数据库;8.对NB公告机构的要求(新法规生效后NB将按照新的要求重新进行授权);9.对高风险器械的新增了要求;MDR&IVDR修订要点15) 欧盟代表与进口商和制造商一起起到连带责任欧盟代表要求, 类似于法规负责人16) 平行贸 Parallel Trade, 特别是重新贴标签或重新包装17) 一次性器械的再加工和使用:- 符合成员国的法规要求- 再加工方要承担法定制造商的责任;- 由机构或外部加工方进行的一次性MD的再加工和再使用必须要符合通用技术规范或协调标准或成员国的法规要求.- 再加工器械的和性能应等同于初期的一次性MD18) 植入器械: 患者应获得基本的信息, 包括标识, 危害健康的警示或注意事项19) 带有CE标记的MD, 在欧盟内可以自由流通和销售. 但成员国可以限制某些器械的使用.20) 除了定制器械外, 所有器械都应应用UDI系统 (MDR执行后1-5年)21) EUDAMed, MD的 命名Code22) 植入和III类产品的制造商应公开产品的主要和性能, 及评估结果的概要.- 和性能的概要应特别包括该产品在与其他诊断或方法比较时的重要性,和二者的使用条件23) 在欧盟的层面上管理NB

SUNGO提醒我们的客户在申请产品CE认证时,在过渡阶段请谨慎考虑是选用法规还是采用老的指令方案,同时也需要对NB机构的发证进行了解和确认以保证产品在欧盟市场销售的可延续性。

MDR适用范围MDR的范围已经扩大了,因此作为制造商,您必须检查您的产品组合,以确定与指令相比,是否有更多的设备属于法规的范围。注意附件十六所列的产品,这些产品一旦通过载列共同规格的实施细则,会纳入本规例。不属于本范围的产品清单见*6段。
我公司申请:出口瑞士:需要瑞士代表,瑞士注册
http://sungoyuan.b2b168.com








