服务欧盟授权代表
代申请欧盟注册
申请CE认证
编写CE技术文件
建立ISO13485
现有MDD客户重大变更的申请期。虽然MDR的正式实施有延期,但是有例外情况如:
· MDD产品的投放截止日期仍为2024年5月26日
· MDD产品供应截止日期仍为2025年5月26日。
欧盟*四版评价(MEDDEV 2.7.1 Rev 4)指南主要变化a)报告更新的频率b)报告编写人和评价人的c)评价报告需要有明确的可测量的目标d)确定技术发展水平e)数据的科学性和有效性f)比对器械g)比对器械的数据获得h)什么时候需要试验i)风险-收益j)售后监督和售后跟踪8)提出Eudamed数据库的建立和使用9)提出器械的可追溯性(UDI)10)对NB提出严格的要求2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
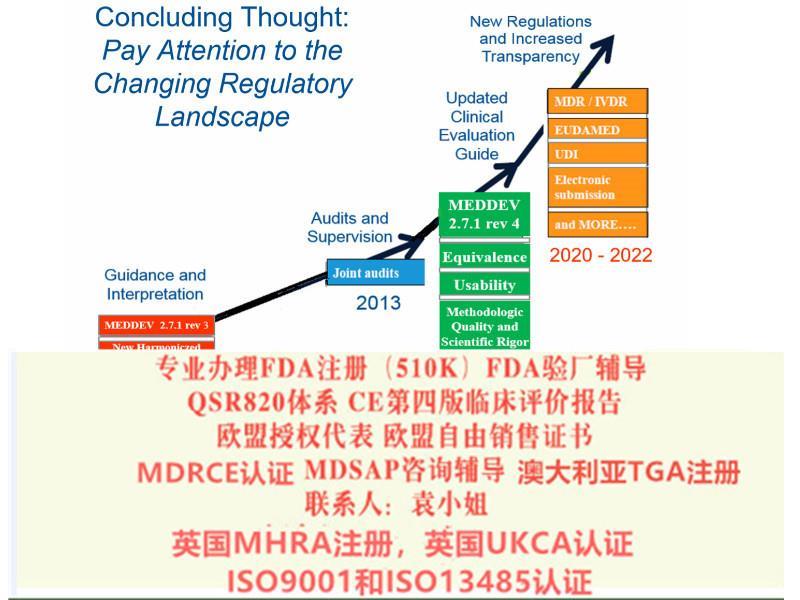
2019 年5 月25 日,MDR 正式生效, 替代了原器械指令(MDD,93/42/EEC)和主动植入式器械指令(AIMD,90/385/EEC)。

2012年,欧盟会起草了MDR和IVDR法规,当中历时了4年的时间,直到2016年6月,欧洲议会和欧盟理事会才发布MDR和IVDR的文本。

我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改
中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。
出口其余国际法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
关于经济运营商各方义务
法规在*I章*2条定义中提出了“经济运营商”的概念,经济运营商是指制造商、授权代表、进口商、经销商以及任何对系统或手术包类器械进行组合或并投放市场的自然人或法人。即在符合法规规定情况下负责器械生产(包括组合或)、销售及上市后运营的自然人或法人。
法规先规定了制造商的义务,涵盖生产、合规、上市后的产品全生命周期,但法规同时规定,经销商、进口商或其他自然人或法人在向市场提供以其名字、注册商标命名的器械时应承担制造商相应的义务,也包括变更相应器械预期用途或变更其他影响其符合性的事项的情况。在上市后要求中,经济运营商同时负有相应的责任和义务。
法规对各方义务的描述更为明确也更为具体,对于制造商的要求更为细化,因此新法规执行后,各方应先明确自身职责和义务,规范有序地开展生产和市场活动,应审核确认上游供应商是否符合规定,并确认能够自己的下游流程符合规定,应按照对应的警戒系统的要求进行或配合事件上报,配合完成现场纠正措施,并依据职责组织培训。
我公司申请: 出口英国需要UKCA认证,英国代表,MHRA注册
http://sungoyuan.b2b168.com








