服务欧盟授权代表
代申请欧盟注册
申请CE认证
编写CE技术文件
建立ISO13485
SUNGO作为**化的器械法规技术服务商,可提供服务:
1. 确定您的器械在美国的具体分类及风险级别;
2. 企业准备510(k)申请所需的资料;
3. 确定检测标准及适用项目,推荐测试机构,对检测报告进行审查;
4. 编撰510(k)申报文件;
5. 企业进行小企业资质申请;
6. 跟踪510(k)文件评审进度;
7. 企业整改发补问题;
8. 进行企业注册和产品列名。
经济运营者是指制造商,授权代表,进口商,分销商以及系统或手术袋的任何组合或。投放市场的自然人或法人。也是说,负责按照法规生产设备(包括组合或),销售和上市后操作的自然人或法人2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于器械*2017/745 号法规(MDR,EU2017/745),5月5日,欧盟(Official Journal of the EuropeanUnion) 正式发布该法规。

*各国转换为法律法规即可实施。在内容方面,MDR基于原始指令的整合,大大改进了器械认证的规范和限制,例如产品分类规则,设备可追溯性,性能研究规范,上市后产品性提高以及有效性方面和。 MDR由10章和123篇文章组成,共17个附录。关于的过渡期:仅拥有根据指令90/385/EEC和93/42/EEC颁发的证书的设备可以投放市场,前提是自MDR应用之日起设计和预期用途没有发生重大变化且符合要求新规定。

我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE*四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
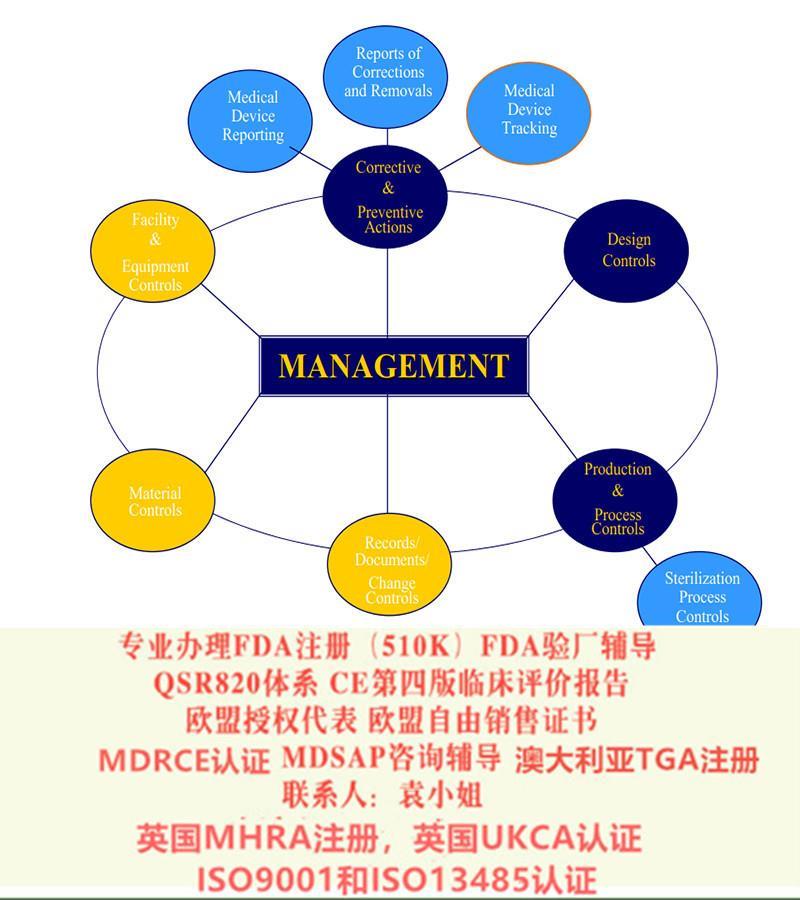
出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改
中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。
出口其余国际法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
2019年5月5日欧盟发布OfficialJournal
在这里需要特别说明的是欧盟此次是直接发布的Regulation(法规)而相比较之前的Directive(指令)其区别在于:提高了文件的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation*向Directive那样需要经过成员国转化成当地法律法规去落实实施。
器械法规(MDR)转换期为3年,2020年5月4日起强制实行。体外诊断器械法规(IVDR)转换期为5年,2022年5月4日起强制实行。MDR将有源器械指令(现行的90/385/EEC)纳入了进来,与一般器械指令(现行93/42/EEC)合二为一,IVDR直接取代了现行的体外诊断器械指令98/79/EEC。
现有指令什么时候停止适用?一般而言,指令90/385 / EEC和93/42 / EEC将于2020年5月26日(DoA)废除。设备和制造商都必须遵守MDR。 您应该评估设备的符合性 - 这可能需要公告机构的参与。其他包括:•评估• 风险管理•质量管理体系(QMS)•上市后监督•技术文档和其他报告•有缺陷设备的责任。在DoA之后,现有指令下的公告机构颁发的证书是否仍然有效?是的,AIMDD / MDD证书通常在的到期日之前有效。 这适用于通知机构通常颁发的所有证书,包括EC设计检验证书,合格证书,EC型式检验证书,EC证书体系和EC证书生产。但是,所有在2017年5月25日之后签发的证书迟在2024年5月27日之前无效。
我公司申请:出口瑞士:需要瑞士代表,瑞士注册
http://sungoyuan.b2b168.com








