服务专业讲解
优势执行力高
价格合理优惠
特点量身定制
周期短
项目ce-mdr
地区全国
标准一站式全包服务
售后服务包售后
Medical Device Regulation 2017/745/EU法规是什么?
SUNGO集团凭借**网络和队伍为**客户提供法规,帮助企业*贸易壁垒,在器械行业尤为专长。
这主要包括:欧盟CE认证(MDD/MDR)、欧盟授权代表、器械欧盟注册、欧盟自由销售证书、FDA注册(FDA510K)、FDA验厂,陪审和翻译、ISO9001/ISO13485,中国局注册证、GMP体系和生产许可证等项目。
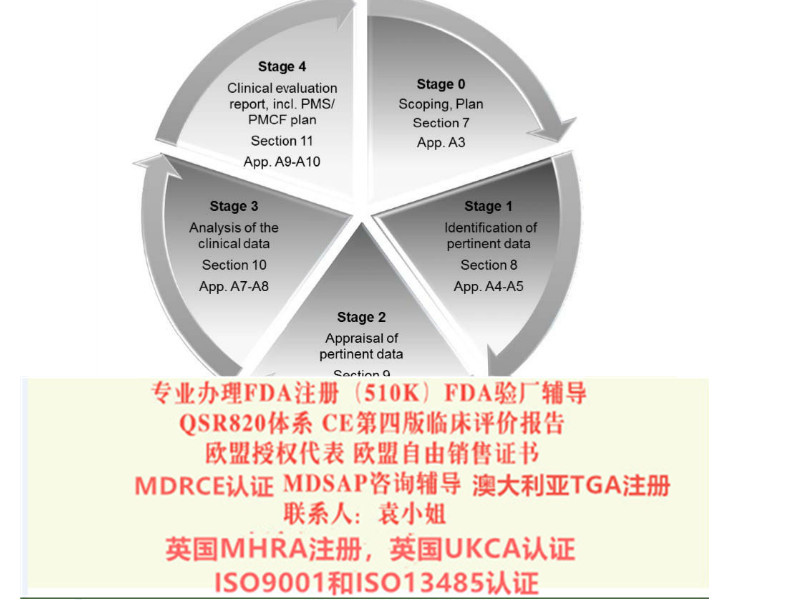
CE*四版评价报告MEDDEV 2.7/1 Rev. 4
近两年,欧盟加强了评价和上市后监督的要求。2016年7月,器械评估的全新文件MEDDEV 2.7/1*4版在欧盟会上正式发布。该指南明确了现有的一些要求,对于制造商如何进行一个健全、系统的评价,以及如何数据和结论的科学有效性有了更明确的。这也给制造商带来了新的挑战。
该指南涉及93/42/EEC和90/385/EEC指令范围内的制造商和公告机构。
该指南包括:明确评估意图、对评估各步骤的详细、明确等同性评估的条件、指出不可用于符合欧洲指令的数据的具体案例、关于评估报告更新的时间安排,对于评估人员资质的低要求、以及此前版本指南不包括的其他具体细节。
公司从2017年年初开展评估业务以来,为国内近两百家器械企业提供了评估报告编撰服务,其中包括很多国内的大型器械企业。*四版欧盟评估指南的发布,的确给企业带来了不小的挑战,此文对主要变化进行了梳理。

器械的制造商有责任进行合格评定,建立技术文件,发布EU合格声明并在产品上粘贴CE标志。这样该产品才能够在EEA市场上交易。
制造商粘贴CE标志的步骤:
确定适用的指令和统一的标准
验证产品特定要求
确定是否需要进行立的合格评定(NB机构参与)
测试产品并检查其合格性
草拟并保留所需的技术文档
贴上CE标志并起草欧盟符合性声明(DOC)
欧洲议会和理事会于2017年4月5日颁布了关于器械的新法规(EU)2017/745,并将在2020年5月26日取代之前的MDD指令和IVDD指令。 ----Regulation (EU) 2017/745 on Medical Devices
2020年4月23 日,欧盟理事会和议会对器械MDR法规进行了修订。规定将MDR法规的实施日期推迟一年,至2021年5月26日。
MDR较之MDD变化主要表现在:
强化制造商的责任:合规负责人/持续更新技术文件/财务**。
更严格的上市前评审:部分产品的分类变高/加强对证据的要求/对特定高风险器械将采用上市前审查机制,由欧盟级别的组参与,进行更严格的事先评估。
适用范围扩大:非用途,但其功能和风险特征与器械相似的器械将同样纳入MDR 的管理范围。
提高透明度和可追溯性:使用器械标识(UDI)系统识别和械/患者将收到具有所有基本信息的植入卡/将建 立包含器械认证信息和研究、警戒和上市后监测信息的修订后的可公开访问的EUDAMED 数据库。

CE技术文件是欧盟器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和,供主管机关抽查,或发生诉讼纠纷时使用。各欧盟指令对于"技术档案"的要求有所差别,在这里谨以中国出口企业常用的“器械”的要求为例,加以说明。
器械MDR法规要求"技术档案"可能包含:企业的质量手册和程序文件;企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的材料),主要内容如下:
1、产品名称、分类
2、产品概述(包括类型和预期用途)
◇产品的历史沿革
◇技术性能参数
◇产品配合使用的附件、配合件和其它设备清单
◇产品的图示与样品
◇产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施
5、生产质量控制
◇产品资料和控制文档(包括产品生产工艺流程图)
◇产品的灭菌方法和确认的描述
◇灭菌验证
◇产品质量控制措施
◇产品稳定性和效期的描述
6、包装和标识
◇包装材料说明
◇标签
◇使用说明书
7、技术评价
◇产品检验报告及相关文献
◇技术概要及观点
8、风险管理
◇产品潜在风险报告及相关文献
◇潜在风险的概要及观点
9、评价
◇产品测试报告及相关文献
◇使用概述及观点
附1、产品出厂检测报告
附2、产品型式检测报告
附3、基本要求检查表
http://sungoyuan.b2b168.com







