2017年5月5日,欧盟正式发布了OfficialJournal其正式对外宣布了新版MDR(REGULATIONEU2017/745)法规和新的IVDR(REGULATIONEU2017/746)法规。新法规将取代现行的三个医疗器械指令:分别是医疗器械指令93/42/EEC,有源医疗器械指令90/385/EEC及体外诊断医疗器械指令98/79/EEC。
我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC药品FDA验厂辅导及整改
中国法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、药监局自由销售证。
出口其余法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
MDR框架
? Chapter I – Scope and Definitions
? Chapter II – CE Marking, Economic Operators, Reprocessing
? Chapter III – Identification and Traceability of Devices
? Chapter IV – Notified Bodies
? Chapter V – Classification and Conformity Assessment
? Chapter VI – Clinical Evaluation and Investigation
? Chapter VII – Vigilance and Market Surveillance
? Chapter VIII – Cooperation between Member States
? Chapter IX – Confidentiality, Data Protection, Funding, Penalties
? Chapter X – Final Provisions
? 100 whereas= Why?
? X Chapters of 123 Articles = What?
? XVI I Annexes = How?
? I General safety and performance requirements
? II Technical documentation
? III Technical documentation on post-market surveillance
? IV EU Declaration of conformity
? V CE marking of conformity
? VI European UDI System
? VII Requirements to be met by notified bodies
? VIII Classification rules
? IX Conformity assessment based on a QMS and assessment of the technical documentation
? X Conformity assessment based on type examination
? XI Conformity assessment based on product conformity verification
? XII Certificates issued by a notified body
? XIII Procedure for custom-made devices
? XIV Clinical evaluation and post-market clinical follow-up
? XV Clinical investigations
? XVI List of groups of products without an intended medical purpose
? XVII Correlation table
? 内容 涉及章节
? 范围&定义 Chapter I
? 产品上市 Chapter II
? UDI & 数据库 Chapter III, Annex VI,XII
? NB Chapter IV , Annex VII,
? 符合性评估 Chapter V, Annex IX, X, XI
? 临床评价和临床调查 Chapter VI, Annex XIV ,XV
? PMS, 警戒系统,上市后监管 Chapter VII , Annex XIV
? Standards, timeline, MDCG Chapter VIII
? Classification Annex VIII
? 一般*性能要求(ER checklist) Annex I
? TD要求 Annex II, III
? DoC &CE marking Annex IV, V
? Custom-made Annex XIII
? 非医疗器械产品清单 & 指令、法规对比表 Annex XVI, XVII

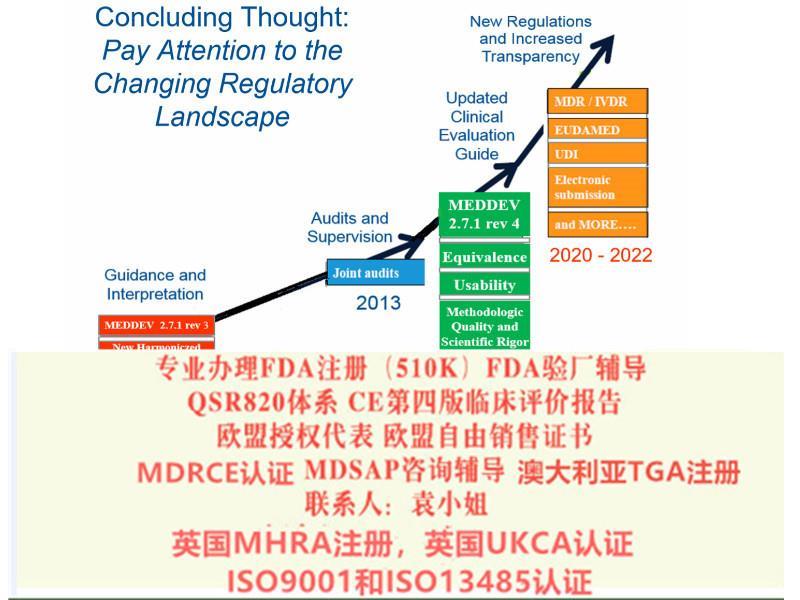
总的说来新的MDR和IVDR加强了体系管理,对高风险设备增加了相关规定比如对于非医疗用途但具有与医疗器械相似特性的设备也将受到新法规的管辖(如用于美容的彩色隐形眼镜),提升了产品对患者的透明度和可追溯性并设立*电子数据库(Eudamed)、医疗设备将有一个一的识别号这加强其在整个供应链的可追溯性。

体外诊断器械法规(IVDR)转换期为5年,2022年5月4日起强制实行。MDR将有源医疗器械指令(现行的90/385/EEC)纳入了进来,与一般医疗器械指令(现行93/42/EEC)合二为一,IVDR直接取代了现行的体外诊断医疗器械指令98/79/EEC。

我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC药品FDA验厂辅导及整改
中国法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、药监局自由销售证。
出口其余法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
MDR&IVDR法规的变化(3)
4. 细化注册要求
General Safety and Performance Requirements
CE技术文件的要求
DOC的要求
NB的授权要求
Implant Card for implanted device
5. 加强市场监管要求
UDI要求
Eudamed数据库
加强欧盟成员国之间的监管协作
NB应满足的要求-Annex VII
? MDR对NB的要求加严格
? NB的授权和监管由多个国家主管当局联合组成的审核小组进行审核;
? NB必须要有自己的审核*团队,新法规下对外聘*的做法将有所限制;
? NB必须要有自己的临床*,而不能仅靠外部临床*进行相关审核。
? 新法规生效后, NB将按照新的资质要求重新授权,不符合要求的NB将会被淘汰。
? 对于已经CE证的企业,应密切关注其NB的新授权以及授权范围!!
与高风险MD有关的新要求
? 对于III类植入器械和用于注入和/或移除药物的IIb类有源器械,其临床评价报告,除了NB审核以外,还需要由NB交至主管当局进行expertpanel review,综合意见进行审评。
? 对于III类和植入器械(顾客定制除外) , 制造商应提交Summary of safety and clinical performance至NB。经NB审核后上传至EUDA MED数据库,供公众查询。
The manufacturer shall mention
on the label or instruc- tions for
use where the summary is available .(Article 32)
? 植入器械- Implant Card
(Article 17)
医疗器械法规(MDR)转换期为3年,2020年5月4日起强制实行。
-/gbaaeee/-
http://sungoyuan.b2b168.com