新MDR对分包方(contract manufacturers)有什么影响?

MDR对动物源性医疗器械的影响以及法规的要求?

办理欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
CE标志有何重要意义
CE标志的意义在于:用CE缩略词为符号表示加贴CE标志的产品符合有关欧洲指令规定的主要要求(Essential Requirements),并用以证实该产品已通过了相应的合格评定程序和/或制造商的合格声明,真正成为产品被允许进入欧共体市场销售的通行证。有关指令要求加贴CE标志的工业产品,没有CE标志的,不得上市销售,已加贴CE标志进入市场的产品,发现不符合*要求的,要责令从市场收回,持续违反指令有关CE标志规定的,将被限制或禁止进入欧盟市场或被迫退出市场。
CE是欧盟强制性的要求,所有销往欧盟市场的产品都必须标示“CE”,当然对作为救死扶伤的医疗器械这一特殊用具而言也不例外。在欧洲,除了主管当局如工商检查者将检查上市的医疗器械是否带有CE标志,海关也将仅允许带有CE标志的产品通过边境。另外,医疗器械的使用者(医生、医院)在购买新器械时也会检查是否带有CE标志。显然,CE标志可作为器械在欧盟内的“通行证”。同时,一个医疗器械产品如果合法加贴了CE标志,也就表明:
1、 该医疗器械符合了欧盟医疗器械法规的基本要求;
2、 该医疗器械可以在欧盟市场内自由流通、销售及使用;
3、 该医疗器械的整个形成过程已通过了一个相应的符合性评价程序。
欧盟医疗器械CE指令
在医疗器械领域,欧盟**发布了三个欧盟指令,以替代原来各成员的认可体系,使有关这类产品投放市场的规定协调一致。 这三个指令分别是:
1.基础医疗器械指令(MDD,93/42/EEC),适用范围很广,包括除有源植入性和体外诊断器械之外的几乎所有的医疗器械,如无源性医疗器械(敷料、一次性使用产品、接触镜、血袋、导管等);以及有源性医疗器械,如核磁共振仪、超声诊断和治疗仪、输液泵等。该指令已于1995年1月1日生效,过渡截止日期为1998年6月13日,从1998年6月14日起强制执行。
2.体外诊断器械指令(IVDD,98/79/EC),适用于血细胞计数器,血糖仪、妊娠检测试纸、**诊断、优生诊断等体外诊断用医疗器械产品。
3.有源植入性医疗器械指令(AIMDD, 90/335/EEC),适用于心脏起搏器,可植入的胰岛素泵等有源植入性医疗器械。AIMD于1993年1月1日生效。过渡截止期为1994年12月31日,从1995年1月1日强制实施。
上述指令规定,在指令正式实施后,只有带有CE标志的医疗器械产品才能在欧盟市场上销售。
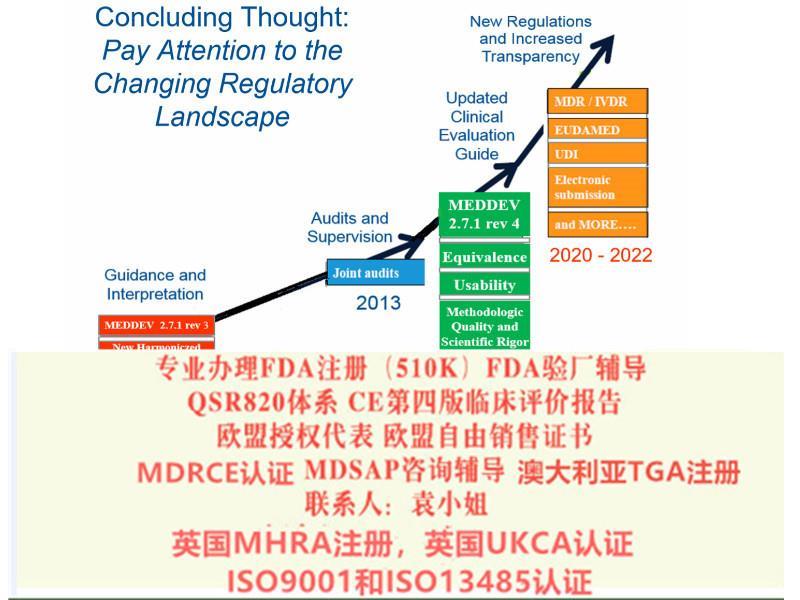
2017 年5 月25 日,MDR 正式生效, 替代了原医疗器械指令
办理欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书
CE技术文件
CE技术文件是欧盟医疗器械指令中很重要的一个事项,它的目的是要求企业准备充份的技术资料和证明,供主管机关抽查,或发生诉讼纠纷时使用。各欧盟指令对于"技术档案"的要求有所差别,在这里谨以中国出口企业较常用的“医疗器械”的要求为例,加以说明。
医疗器械指令93/42/EEC要求"技术档案"可能包含:企业的质量手册和程序文件;企业简介及欧洲代表名称、联系方式;CE符合性声明(或称自我保证声明,若该产品是和其它设备联合运用,则应有整体符合基本要求的证明材料),主要内容如下:
1、产品名称、分类
2、产品概述(包括类型和预期用途)
◇产品的历史沿革
◇技术性能参数
◇产品配合使用的附件、配合件和其它设备清单
◇产品的图示与样品
◇产品所用原材料及供应商
3、使用该产品的调和标准/或其它标准
4、风险分析评估结论和预防措施
5、生产质量控制
◇产品资料和控制文档(包括产品生产工艺流程图)
◇产品的灭菌方法和确认的描述
◇灭菌验证
◇产品质量控制措施
◇产品稳定性和效期的描述
6、包装和标识
◇包装材料说明
◇标签
◇使用说明书
7、技术评价
◇产品检验报告及相关文献
◇技术概要及*观点
8、风险管理
◇产品潜在风险报告及相关文献
◇潜在风险的概要及*观点
9、临床评价
◇产品临床测试报告及相关文献
◇临床使用概述及*观点
附1、产品出厂检测报告
附2、产品型式检测报告
附3、基本要求检查表
备注:
◇临床研究(包括:物理性能,生化、药理 、药动及毒性研究,功效测试,灭菌合格证明,药物相容性等)
◇生物兼容性测试(A)一部分要求:细胞毒性、感光性、刺激-皮内反应、急性全身中毒、致热性、亚急性中毒、遗传毒性、植入溶血性; (B)支持测试:慢性中毒、致癌性、再生性/生长性毒素、生物动因退化。)
◇临床资料(需要临床研究或描述临床研究)
◇包装合格证明
◇标签、使用说明
◇结论(设计档案资料的接受、利益对应风险的陈述)
10、欧盟授权代表信息及协议
11、符合基本要求表
12、协调标准
13、警戒系统程序
MDR 由指令升级为法规,提高了对欧盟成员国的约束力
2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于医疗器械2017/745 号法规
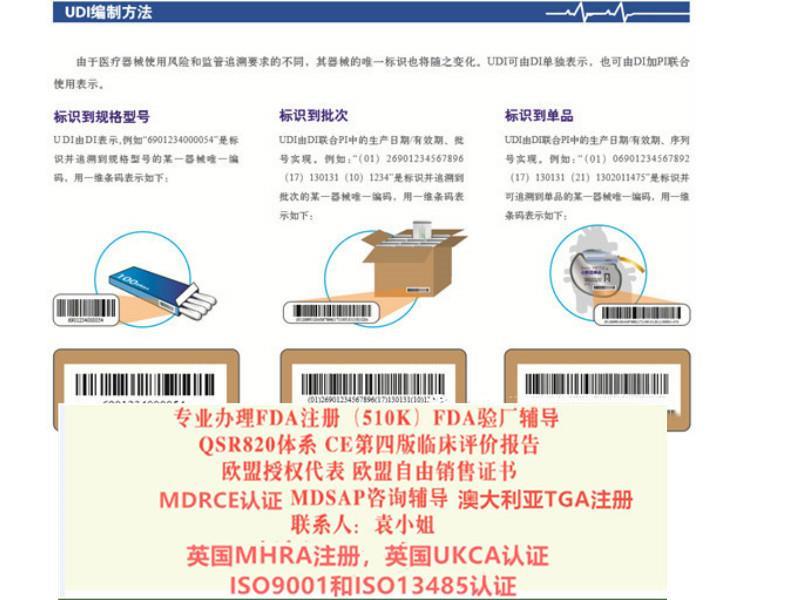
-/gbaaeee/-
http://sungoyuan.b2b168.com