我们的美国FDA代理人服务包括:我们能提供合理的收费,尽量减少您的不必要的开支
我们可以为您提供的自主服务项目主要有:
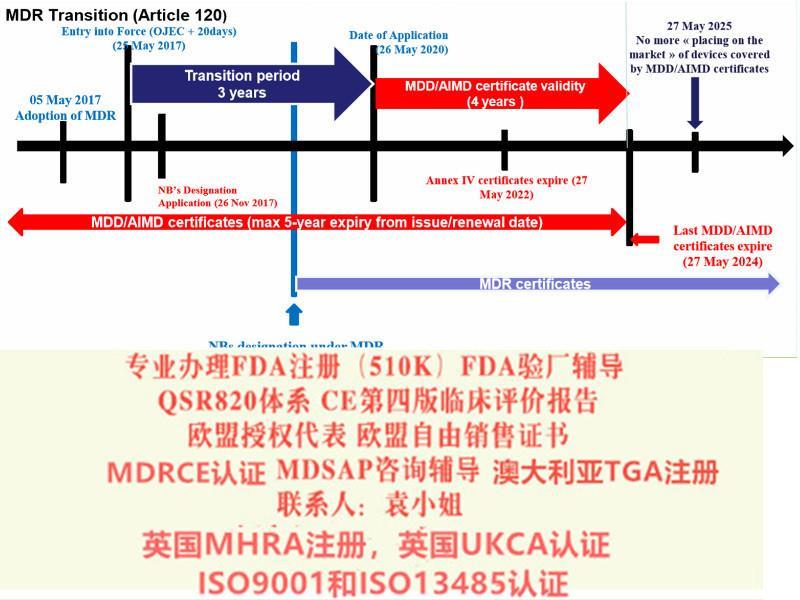
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC药品FDA验厂辅导及整改
中国法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、药监局自由销售证。
出口其余法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
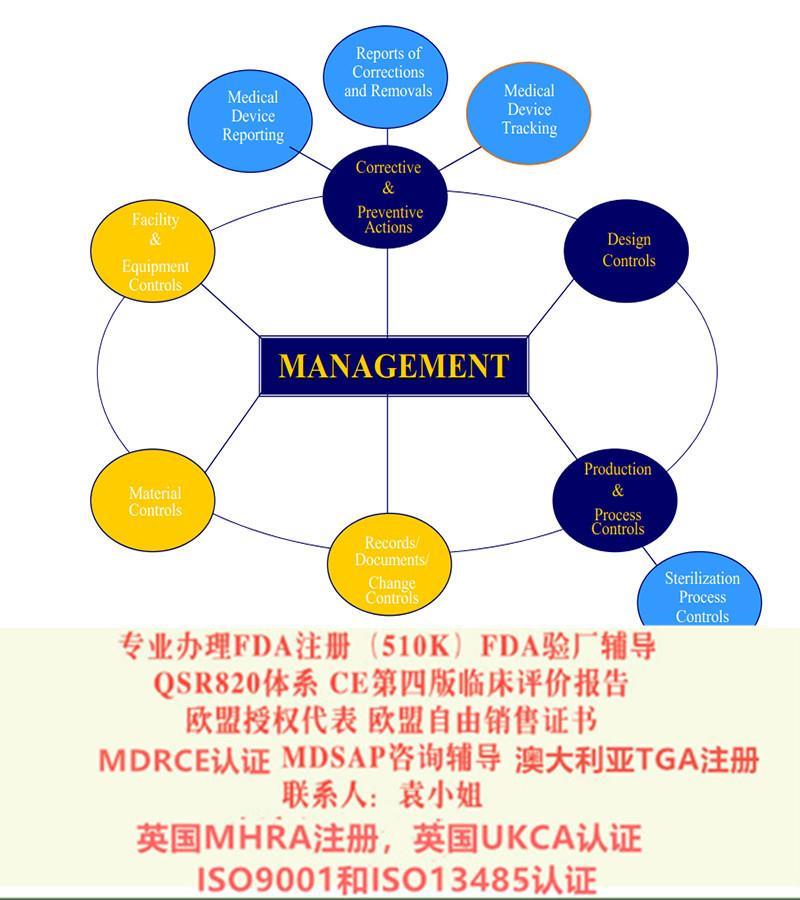
FDA 验厂
FDA是美国食品和药物监督管理局(Food and Drug Administration)的简称,是美国**在健康与人类服务部(Department Of Health and Human Services) 和公共卫生部(Public Health Service)中设立的执行机构之一,其主要主管:食品、药品、医疗器械、食品添加剂、化妆品、及药品等产品的监督检验。跟据规定,上述产品必须经过FDA检验证明*后,方可在市场上销售。当然由于医疗器械本身的特殊性,FDA也常与职业卫生与*署(Occupational Health and Safety Administration)、美国海关(U.S.CustomsService)及核能管理**(Nuclear Regulatory Commission)等其他部门协调合作FDA下属的CDRH(器械与放射健康中心)是专职负责医疗器械企业管理的**机构,其根据FDA的授权,安排检查员到各企业进行工厂检查。一般美国境内企业:一般每两年检查一次;美国境外企业:不定期检查;FDA官方所有检查费用由FDA承担。FDA检查官,有些是直接从美国过来的,有一些,是FDA中国办公室的人员。中国现有三个办公室,上海,北京,广州。中国办事处可以进行审核的,但审核任务仍由FDA总部分配。随员的不同,具体的做法有较大差异。审核的侧重点,会与检查官的个人经历、知识都有一定的关系。
审厂的触发原因包括:
一,例行检查;
二、FDA需要调查行业数据;
三,发生顾客抱怨,特别是多次的抱怨
四、发生较多的不良事件;
五,产品多次出现质量问题;
六、FDA接受其他管理局的委托进行审厂。比如,接受退伍军人管理局的委托。也可以主动申请审厂。有时候,你的美国客户,也可以申请FDA来审厂。审查不通过,只要后续措施得力,通常不会导致罚款之类的处罚。对审查的结果,应严格按照审核官的要求,提供详细的整改资料,不会立即停止销售。当然,有的整改措施是需要效果验证的。你就需要同时提交效果验证的证据。审厂不通过,不会直接导致罚款。
我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:医疗器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂辅导及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系辅导、食品FDA验厂辅导及整改、OTC药品FDA验厂辅导及整改
中国法规:医疗器械产品备案登记表、医疗器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证辅导、SFDA验厂辅导、SFDA注册检测、企业标准编制、药监局自由销售证。
出口其余法规:医疗器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂辅导、ISO22716 GMPC验厂辅导、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
FDA官方对于美国代理人的解释也可以参考看一下的原文:
Any foreign establishment engaged in the manufacture, preparation, propagation, compounding, or processing of a device imported into the United States must identify a United States agent (U.S. agent) for that establishment.
Information about a foreign establishment’s U.S. Agent is submitted electronically using the FDA Unified Registration and Listing System (FURLS system) and is part of the establishment registration process. Each foreign establishment may designate only one U.S. agent. The foreign establishment may also, but is not required to, designate its U.S. agent as its official correspondent. The foreign establishment should provide the name, address, telephone and fax numbers, and e-mail address of the U.S. agent.
The U.S. agent identified will be required to complete an automated process to confirm that they have agreed to act as the U.S. agent. The automated process will forward an email verification request to the U.S. agent. They will be requested to confirm her/his consent to act as a representative/liaison on behalf of the foreign establishment. If the U.S. agent denies consent (or does not respond within 10 business days), the Official Correspondent/Owner Operator of the foreign establishment will be notified and must designate a new U.S. agent to satisfy the regulatory obligation.
Responsibilities of a U.S. agent
The U.S. agent must either reside in the U.S. or maintain a place of business in the U.S. The U.S. agent cannot use a post office box as an address. The U.S. agent cannot use just an answering service. They must be available to answer the phone or have an employee available to answer the phone during normal business hours.
The responsibilities of the U.S. agent are limited and include:
assisting FDA in communications with the foreign establishment,
responding to questions concerning the foreign establishment's devices that are imported or offered for import into the United States,
assisting FDA in scheduling inspections of the foreign establishment and
if FDA is unable to contact the foreign establishment directly or expeditiously, FDA may provide information or documents to the U.S. agent, and such an action shall be considered to be equivalent to providing the same information or documents to the foreign establishment.
Please note that the U.S. agent has no responsibility related to reporting of adverse events under the Medical Device Reporting regulation (21 CFR Part 803), or submitting 510(k) Premarket Notifications (21 CFR Part 807, Subpart E).

-/gbaaeee/-
http://sungoyuan.b2b168.com








