项目背景()
欧洲议会和理事会于2017 年4 月5 日签发的关于医疗器械2017/745 号法规, 修订了2001/83/EC 号指令,178/2002 号(EU)法规和1223/2009 号(EU)法规,并废除了理事会90/385/EEC 号和93/42/EEC 号指令.
MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性。依据Article 120 clause2 的规定,过渡期内NB签发的CE证书继续有效。至2020年5月26日,MDR法规将*强制实施。至2022年5月26日IVDR法规将*强制实施。至2014年,MDD/ AIMD证书全部失效。
MDR新法规变化
1)扩大了应用范围
2)提出了新的概念和器械的定义
3)细化了医疗器械的分类
4)完善了器械的通用*和性能要求
5)加强对技术文件的要求
6)加强器械上市后的监管
7)完善临床评价相关要求
事故对于患者意味着伤害,对于企业的生存也具有巨大的破坏力。因此,临床评估的设计和临床数据的收集具有至关重要的意义。然而在实践中,许多制造商不清楚什么是欧盟法规所要求的临床评估,什么样的临床数据能满足欧盟的法规要求。
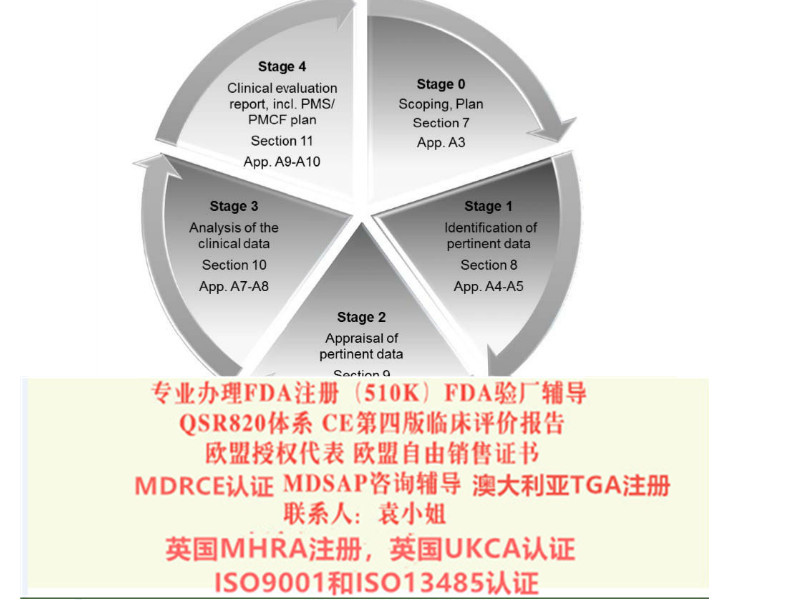
欧盟四版临床评价(MEDDEV 2.7.1 Rev 4)指南主要变化
a)临床报告新的频率
b)报告编写人和评价人的
c)评价报告需要有明确的可测量的目标
d)确定技术发展水平
e)数据的科学性和有效性
f)比对器械
g)比对器械的数据获得
h)什么时候需要临床试验
i)风险-收益
j)售后监督和售后临床跟踪
8)提出Eudamed数据库的建立和使用
9)提出器械的可追溯性(UDI)
10)对NB提出严格的要求

解读MDR系列讨论 部分:MDR演变过程和MDR的过渡期
MDR演变过程
近两年,国内外医疗器械法规变化巨大,对于医疗器械厂家及从业人员来说,都面临着比较大的挑战,针对明年将要*实施的欧盟医疗器械新法规2017/745/EU MDR,小编精心为大家准备了解读MDR系列,旨在为从业的朋友们提供有价值的参考信息。
今天给大家带来解读MDR系列讨论 部分:演变过程和MDR的过渡期。
众所周知,现行的MDD 医疗器械指令93/42/EEC是1993颁布的,距今已经有26年的历史,这期间,医疗器械行业无论是从技术方面,应用方面都有了巨大的变革,无疑,一部用了26年的指令已经过于陈旧,新法规替代老法规已经势在必行。2010年发生的法国PIP事件也促使了欧盟推行欧盟医疗新法规的起草和推行。
2012年,欧盟会起草了MDR和IVDR法规,当中历时了4年的时间,直到2016年6月,欧洲议会和欧盟理事会才发布MDR和IVDR的文本。

医疗器械法规(MDR)何时适用
MDR(EU)2017/745将于2020年5月26日起申请 - “申请日期”(DoA)。
MDR的某些条款将提前生效(例如关于公告机构和医疗器械协调组)。 有些将在稍后申请(例如关于UDI标签)。
现有指令什么时候停止适用?
一般而言,指令90/385 / EEC和93/42 / EEC将于2020年5月26日(DoA)废除。
设备和制造商都必须遵守MDR。 您应该评估设备的符合性 - 这可能需要公告机构的参与。
其他重点包括:
•临床评估
• 风险管理
•质量管理体系(QMS)
•上市后监督
•技术文档和其他报告
•有缺陷设备的责任。

一,DMR的主要变化
1.扩大了应用范围
2.提出了新的概念和器械的定义
3.细化了医疗器械的分类
4.完善了器械的通用*和性能要求
5.加强对技术文件的要求
6.加强器械上市后的监管
7.完善临床评价相关要求
8.提出Eudamed数据库的建立和使用
9.提出器械的可追溯性(UDI)
10.对NB提出严格的要求
MDR简介
2017年5月5日,欧盟期刊(Official Journal of the European Union)正式发布了欧盟医疗器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类医疗器械指令)and 93/42/EEC(医疗器械指令)。依据MDR Article 123的要求,MDR于2017年5月26日正式生效,并与2020年5月26日期正式取代MDD(93/42/EEC)和AIMDD(90/385/EEC)。
医疗器械食品化妆品法规服务
MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性。依据Article 120 clause2 的规定,过渡期内NB签发的CE证书继续有效,但是从其交付日期起有效期不**过5年,并且于2024年5月27日失效。
-/gbaaeee/-
http://sungoyuan.b2b168.com