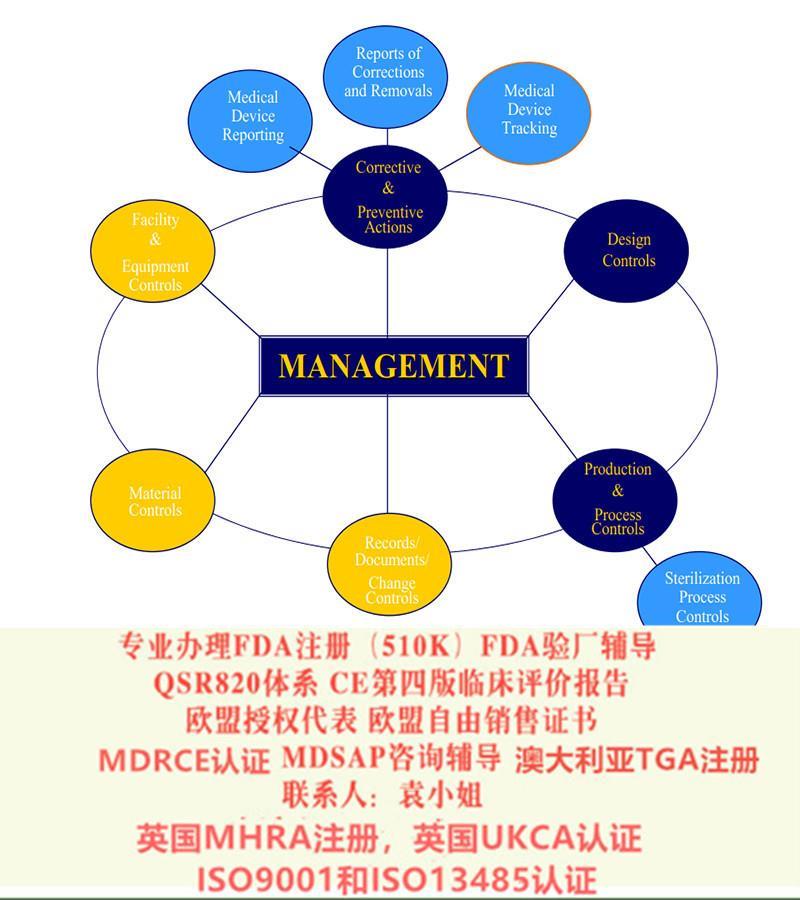
我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改
中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。
出口其余法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
经销商:
- 应确认产品上已正确使用CE标志、制造商已经签署符合性声明、标签说明书语言符合法规要求、已正确使用UDI;
- 应确认产品包装或其随机文件上有进口商的信息;
- 应确保器械在符合生产商要求的环境里存储和运输;
- 应对客户投诉、召回、不合格品记录在案,并通知生产商及欧盟代表;
- 认为产品不符合MDR&IVDR要求的,应立即通知制造商或欧盟代表或进口商;如果涉及严重风险的,应立即通知主管当局。
UDI-器械识别码
UDI是Unique Device Identification的缩写,是显示在产品标签上的一组条形码或二维码,由器械识别码DI和生产识别码PI组成,前者包括制造商和产品名称信息,后者包括产品批号、有效期等信息;
制造商发货、进口商验收、销售商收货时都需要扫描UDI,
数据在UDI数据库中保存,从而实现产品全程可追溯;
UDI中的Basic UDI-DI也将出现在
制造商签发的CE符合性声明中,
也会显示在CE证书上;
欧盟授权GS1、HIBCC、ICCBBA
这三家美国公司管理UDI,制造
商应自行向三家机构申请产品的
我们可以为您提供的自主服务项目主要有:
出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证
出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改
中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。
出口其余法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试
MDR范围的变化
MDR纳入多产品进行管理;
• MDR中附录XIV所列的非用途的产品:
• 隐形眼镜;
• 用于修改或固定身体部位的植入物;
• 面部或其他皮肤或粘膜填充剂;
• *设备;
• 用于人体的有创激光设备;
• 强脉冲光设备
MDR分类规则的主要变化
• 仍为4类: Class I, IIa, IIb, III
• Duration of Use (无变化)
-Transient < 60 minutes;
- 60minutes < Short-term<30 days;
- Long term > 30 days
• 18 个规则=> 22个规则
- Rule 1-4 Non invasive devices
- Rule 5-8 Invasive devices
- Rule 9-12 Active devices
- Rule 13-22 Special rules
分类规则: 1-4 Non invasive devices
Rule 3:
• 在原来基础上, 新增由一种或几种物质混合组成的非侵入器械,其预期用于直接在体外接触已从人体或人体胚胎中取出且还未被植入人体的细胞、组织或,该类器械属于III类。
分类规则: 5-8 Invasive devices
Rule 6、 7:
• 在原来基础上, 新增用于直接接触心脏或循环系统的短暂、短期使用型侵入性手术器械属于III类

办理出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认证(CE整套技术文件编订、 CE四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟自由销售证书、英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。

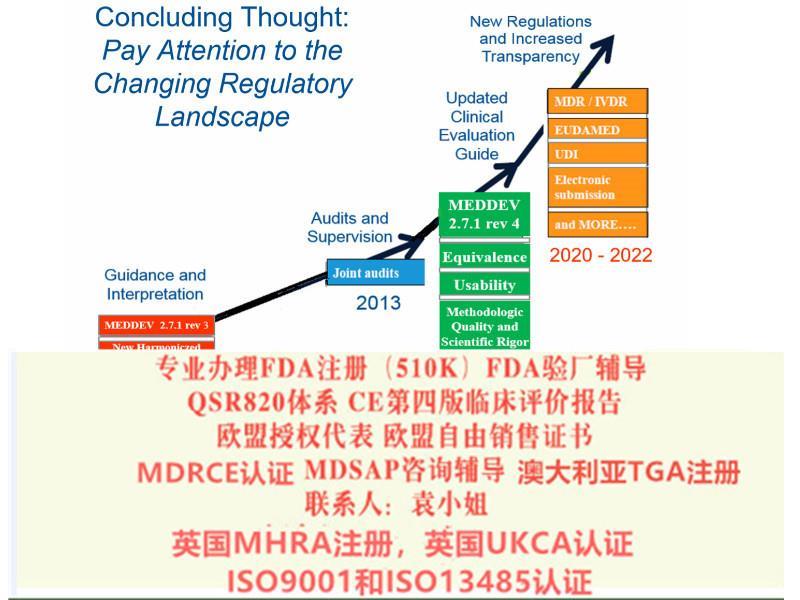
2019年2月器械法规(MDR)和体外诊断器械法规(IVDR)终提案发布,2019年3月7日欧盟28个成员国一致表决同意欧盟采用新版的器械法规(MDR)和体外诊断器械法规(IVDR)。
http://sungoyuan.b2b168.com